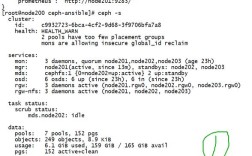
在使用R语言进行生物信息学分析时,报错是一个常见问题,这些错误可能由于多种原因引起,包括数据格式不正确、代码语法错误、包或函数使用不当等,本文将通过案例分析,帮助读者理解报错原因,并提供解决方案。
常见报错及解决方案
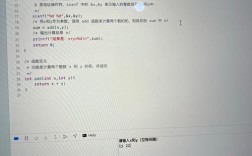
1、DESeq包安装和加载问题:

报错描述:在尝试安装和加载DESeq包时,出现以下报错信息:“Warning: namespace ‘DESeq’ is not available and has been replaced by .GlobalEnv when processing object ‘cds’”和“Error in .requirePackage(package) : unable to find required package ‘DESeq’”。
解决方案:确保已正确安装并加载了BiocManager包,这是用于管理Bioconductor包的工具,使用以下命令尝试重新安装DESeq包:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("DESeq")如果仍然无法解决问题,请检查R版本是否与DESeq包兼容,有时,可能需要更新R版本或寻找适用于当前R版本的DESeq包版本。
2、数据格式问题:
报错描述:在使用DESeq包进行差异表达分析时,代码运行出错。
解决方案:确保输入的数据格式符合要求,DESeq包通常要求输入的数据格式为data.frame或list,还需要确保基因表达矩阵和样本信息矩阵的列名匹配。

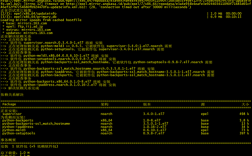
3、依赖包缺失问题:
报错描述:在安装DESeq包时,出现缺少locfit包或xtable包的报错信息。
解决方案:确保已安装所有必要的依赖包,可以通过以下命令安装缺失的包:
install.packages("locfit")
install.packages("xtable")FAQs
1、为什么在尝试安装DESeq包时会出现“package ‘DESeq’ is not available for this version of R”的警告?
DESeq包可能不适用于当前的R版本,建议检查R版本是否与DESeq包兼容,或者寻找适用于当前R版本的DESeq包版本。
2、如何解决“namespace ‘DESeq’ is not available and has been replaced by .GlobalEnv when processing object ‘cds’”的问题?

这通常是因为DESeq包尚未正确安装或加载,按照上述解决方案中的步骤重新安装和加载DESeq包即可解决此问题。
3、在使用DESeq包进行差异表达分析时,还需要注意哪些事项?
确保输入的数据已经过适当的预处理,例如过滤掉低表达量的基因、去除批次效应等。
根据实验设计选择合适的模型参数,例如dispersion估计方法、正规化方法等。
对结果进行适当的解释和可视化,以便于生物学意义的解读。
通过以上分析和解答,希望能够帮助您解决在使用R语言进行生物信息学分析时遇到的DESeq包报错问题,如果问题仍然存在,请考虑寻求专业的技术支持或查阅相关文档和论坛以获取更多帮助。